用R语言进行细胞流式分析及作图
首先进行依赖包的安装
install.packages("BiocManager")
install.packages("devtools")
BiocManager::install("cytolib") #不安装这个依赖包后续安装中会出错
BioManager::install("flowCore") #用于后缀为.fcs的文件
BiocManager::install("flowViz") #基础的可视化工具
BiocManager::install("ggcyto") #使用ggplot命名法的高级可视化工具
BiocManager::install("openCyto") #用于链接各种分析方法
BiocManager::install("flowWorkspace") #用于建立分析模板
BiocManager::install("CytoML") #插入 FlowJo and DiVA 工作区
BiocManager::install("flowAI")
BiocManager::install("flowClean")
BiocManager::install("flowCut") #这些依赖包很大程度上需要彼此工作(除了flowCore,它是“基础包”)。so文件经常会在没有人为操作的情况下互相加载。为简单起见,我们将它们全部加载。您可能需要“清理”数据。flowaAI 和 flowCut是我的推荐。其中flowClean是原始的,但被flowCut取代。因此安装以下依赖包。
devtools::install_github("DillonHammill/CytoExploreRData")
devtools::install_github("DillonHammill/CytoExploreR",build_vignettes = TRUE) #一个有趣的依赖包是CytoExploreR,它试图将 R 的强大功能与鼠标的易用性结合起来。可以可视化的进行编辑和做图。
#这两个依赖包可能有点大,第一个包有3GB,建议使用断点续存的方式进行下载后在安装依赖。整体的安装过程可能有些枯燥和耗时,需要耐心等待。加载需要的依赖包
library(flowCore)
library(flowAI)
library(ggcyto)
library(ggplot2)
fs <- read.FCS(filename = "/mnt/chromeos/MyFiles/Downloads/debian/flow_Cytometry/Sample/0001.FCS", alter.names=TRUE) #将需要的文件在R语言中打开
fs_comp <- compensate(fs, spillover(fs)$SPILL) #补偿表也可以在Flowjo软件中进行修改使图像更接近于真实水平
fs_comp_clean <- flow_auto_qc(fs_comp)## Quality control for the file: 8c0a4953-f1da-4134-880e-4918967e16cb
## 14.21% of anomalous cells detected in the flow rate check.
## 0% of anomalous cells detected in signal acquisition check.
## 0.09% of anomalous cells detected in the dynamic range check.#接下来,用flow_auto_clean()的命令清理文件中流式数据的细胞碎片和死细胞
fs_comp_clean #输出清理后的数据## flowFrame object '8c0a4953-f1da-4134-880e-4918967e16cb'
## with 428572 cells and 11 observables:
## name desc range minRange maxRange
## $P1 FSC.A NA 262144 0.00 262144
## $P2 SSC.A NA 262144 -111.00 262144
## $P3 FITC.A IFNa 262144 -63.45 262144
## $P4 PE.A CD123 262144 -101.25 262144
## $P5 PerCP.Cy5.5.A MHCII 262144 -75.60 262144
## $P6 PE.Cy7.A CD14 262144 -99.90 262144
## $P7 APC.A CD11c 262144 -48.10 262144
## $P8 APC.Cy7.A IL6 262144 -110.50 262144
## $P9 Pacific.Blue.A IL12 262144 -83.70 262144
## $P10 Alex.700.A TNFa 262144 -48.10 262144
## $P11 Time NA 262144 0.00 262144
## 175 keywords are stored in the 'description' slotfs #与清理前的数据进行对比## flowFrame object '8c0a4953-f1da-4134-880e-4918967e16cb'
## with 500000 cells and 11 observables:
## name desc range minRange maxRange
## $P1 FSC.A NA 262144 0.0 262143
## $P2 SSC.A NA 262144 -111.0 262143
## $P3 FITC.A IFNa 262144 -111.0 262143
## $P4 PE.A CD123 262144 -111.0 262143
## $P5 PerCP.Cy5.5.A MHCII 262144 -111.0 262143
## $P6 PE.Cy7.A CD14 262144 -111.0 262143
## $P7 APC.A CD11c 262144 -80.6 262143
## $P8 APC.Cy7.A IL6 262144 -111.0 262143
## $P9 Pacific.Blue.A IL12 262144 -111.0 262143
## $P10 Alex.700.A TNFa 262144 -75.4 262143
## $P11 Time NA 262144 0.0 262143
## 175 keywords are stored in the 'description' slot#显然剔除了一些细胞碎片和死细胞,可以使流式的散点图更规整接着,转换文件并作图
trans <- estimateLogicle(fs_comp_clean, colnames(fs_comp_clean[,3:10]))
fs_comp_clean_trans <- transform(fs_comp_clean, trans)
ggcyto(fs_comp_clean_trans,aes(x="FITC.A",y="PE.A")) +
geom_hex(bins=256) +
theme_classic() #成功输出了流式散点图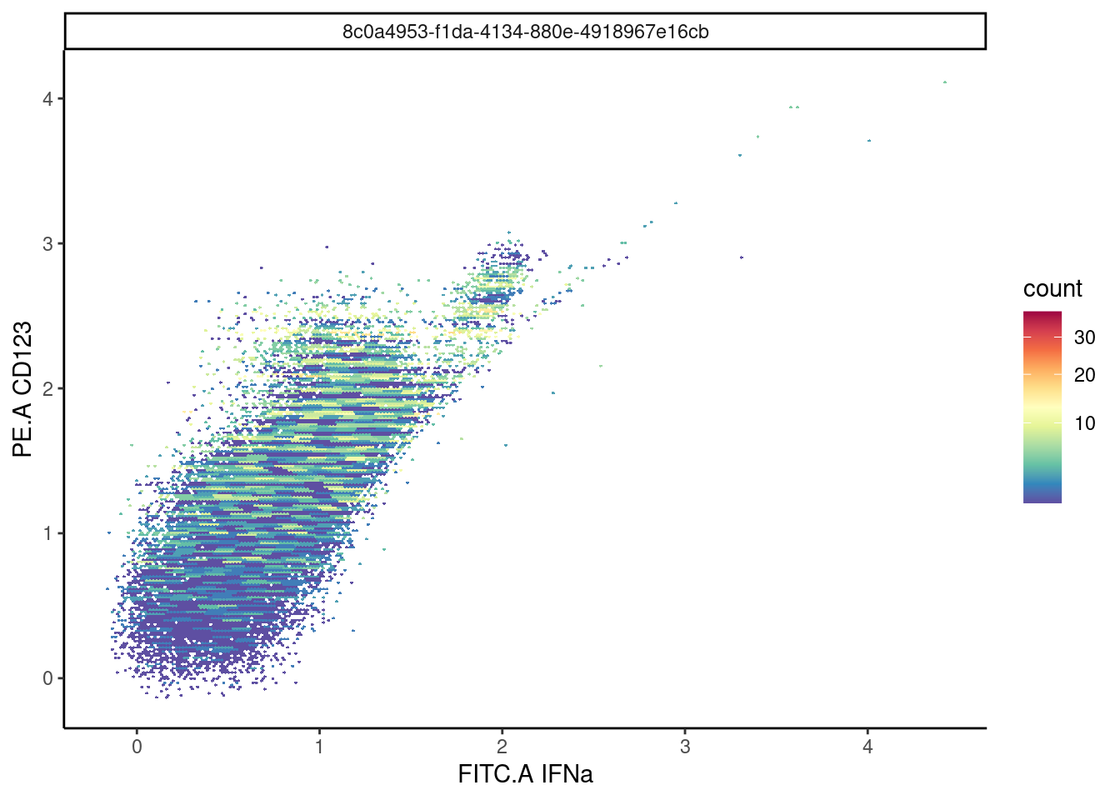
ggcyto(fs_comp_clean_trans,aes(x="FITC.A")) +
geom_density() +
theme_classic() #成功输出了密度图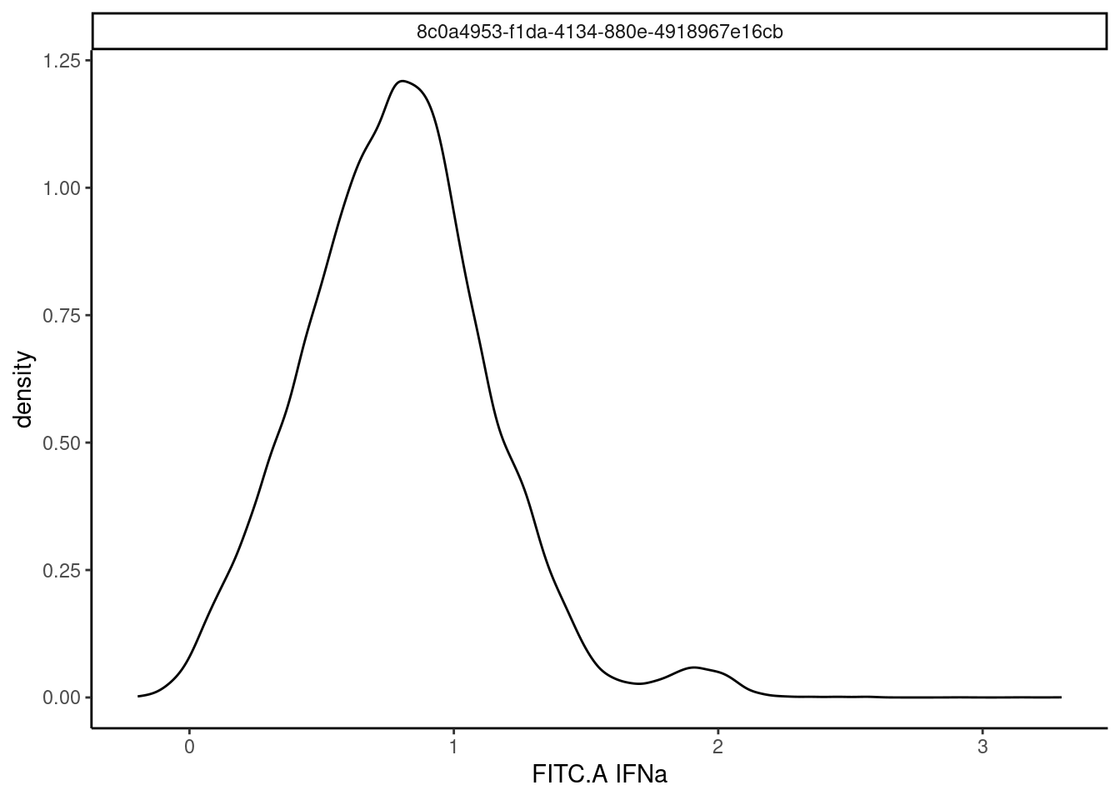
比如说在之前的作图中,我们发现按照教程做出来的图的横纵坐标都非常小。列位可能会有疑惑:难道是依赖包的自动清理死细胞和细胞碎片的问题吗?
经过一些尝试和考察,小编发现:其原因是在于以下命令行
trans <- estimateLogicle(fs_comp_clean, colnames(fs_comp_clean[,3:10]))
fs_comp_clean_trans <- transform(fs_comp_clean, trans)
fs_comp_clean## flowFrame object '8c0a4953-f1da-4134-880e-4918967e16cb'
## with 428572 cells and 11 observables:
## name desc range minRange maxRange
## $P1 FSC.A NA 262144 0.00 262144
## $P2 SSC.A NA 262144 -111.00 262144
## $P3 FITC.A IFNa 262144 -63.45 262144
## $P4 PE.A CD123 262144 -101.25 262144
## $P5 PerCP.Cy5.5.A MHCII 262144 -75.60 262144
## $P6 PE.Cy7.A CD14 262144 -99.90 262144
## $P7 APC.A CD11c 262144 -48.10 262144
## $P8 APC.Cy7.A IL6 262144 -110.50 262144
## $P9 Pacific.Blue.A IL12 262144 -83.70 262144
## $P10 Alex.700.A TNFa 262144 -48.10 262144
## $P11 Time NA 262144 0.00 262144
## 175 keywords are stored in the 'description' slot在这个命令中,存在一些问题在于实验目的,比如我们实验中只做了一个FITC的单染,并没有做其他染色。要如何绘制流式散点图呢?比如:常见到的sci中的横纵坐标为科学计数法的形式。则整体应该修改代码
trans <- estimateLogicle(fs_comp_clean, colnames(fs_comp_clean[,4]))
fs_comp_clean_trans <- transform(fs_comp_clean, trans)
#在转换中只留FITC的信号而屏蔽其他荧光区的杂信号。
ggcyto(fs_comp_clean_trans,aes(x="FITC.A",y="PE.A")) +
geom_hex(bins=256) +
labs(x="FITC-A", y="PE-A") +
scale_x_flowjo_biexp() +
theme_classic()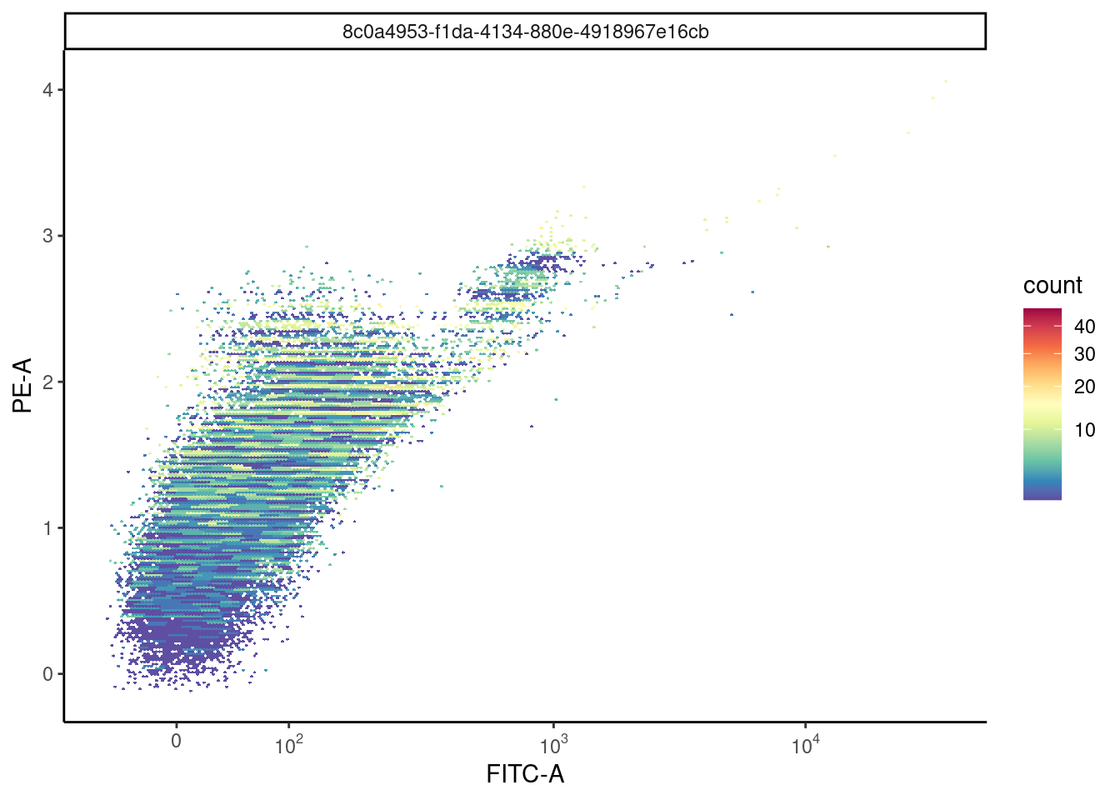
在分析及绘图中仍然存在一些问题,有一些是依赖包自身的问题,有一些是R语言版本的问题,小编会在接下来的文章中陆续解决这些问题。
比如说,流式的原始数据不可能只有一个,我们需要将空白、阴性对照、阳性对照及实验组同时加载进来,同时进行分析处理。
在整个的flowSet中需要输入以上命令进行查看其中某一个数据。其中有几项命令是需要用空白去补偿和转换的,则需要调用blank的文件。
myfiles <- list.files(path="/mnt/chromeos/MyFiles/Downloads/debian/flow_Cytometry/Sample/", pattern=".FCS")
fs <- read.flowSet(myfiles[1:3], path="/mnt/chromeos/MyFiles/Downloads/debian/flow_Cytometry/Sample/", alter.names = TRUE)
fs[[1]]## flowFrame object '0001.FCS'
## with 500000 cells and 11 observables:
## name desc range minRange maxRange
## $P1 FSC.A NA 262144 0.0 262143
## $P2 SSC.A NA 262144 -111.0 262143
## $P3 FITC.A IFNa 262144 -111.0 262143
## $P4 PE.A CD123 262144 -111.0 262143
## $P5 PerCP.Cy5.5.A MHCII 262144 -111.0 262143
## $P6 PE.Cy7.A CD14 262144 -111.0 262143
## $P7 APC.A CD11c 262144 -80.6 262143
## $P8 APC.Cy7.A IL6 262144 -111.0 262143
## $P9 Pacific.Blue.A IL12 262144 -111.0 262143
## $P10 Alex.700.A TNFa 262144 -75.4 262143
## $P11 Time NA 262144 0.0 262143
## 176 keywords are stored in the 'description' slotspillover(fs[[1]])## $SPILL
## FITC.A PE.A PerCP.Cy5.5.A PE.Cy7.A APC.A APC.Cy7.A Pacific.Blue.A
## [1,] 1 0 0 0 0 0 0
## [2,] 0 1 0 0 0 0 0
## [3,] 0 0 1 0 0 0 0
## [4,] 0 0 0 1 0 0 0
## [5,] 0 0 0 0 1 0 0
## [6,] 0 0 0 0 0 1 0
## [7,] 0 0 0 0 0 0 1
## [8,] 0 0 0 0 0 0 0
## Alex.700.A
## [1,] 0
## [2,] 0
## [3,] 0
## [4,] 0
## [5,] 0
## [6,] 0
## [7,] 0
## [8,] 1
##
## $spillover
## NULL
##
## $`$SPILLOVER`
## NULLfs_comp <- compensate(fs[[1]], spillover(fs[[1]])$SPILL)
##或者用以下命令
comp <- fsApply(fs, function(x)spillover(x)[[1]], simplify=FALSE)
fs_comp <- compensate(fs, comp)
##其中还有转换命令与之不同
trans <- estimateLogicle(fs_comp[[1]], colnames(fs_comp[[1]][,5:10]))
fs_comp_trans <- transform(fs_comp, trans)其中需要再提及的是一般而言,在这两种命令后输出的都是flowSet格式的数据,其方便的地方在于后续建立门(gate)
若其中fs_comp_trans不为flowSet格式,即不是单一的fcs文件,则可以建立门。若为单一文件则会出现报错。
另一方面,在多个文件进行绘图时也会出现一些问题,在于绘图过程中,会自动显示facet框架,其中会显示文件名+后缀的情况。这种需要我们进行手动的编辑
我们需要修改他们的名字
##用以下命令修改名字
fs_comp_trans@phenoData@data$name[1] <- "Blank"
pData(fs_comp_trans)## name
## 0001.FCS Blank
## 0002.FCS 0002.FCS
## 0003.FCS 0003.FCS这个细节在作图中其实是至关重要的。
在细胞流式技术中,最关键的地方在于门(gate)的建立,若门的范围选择的不对的话,显示的荧光强度和细胞种类都会出现问题
rg1 <- rectangleGate("FSC.A"=c(25000, Inf), filterId = "NoneDebris")
gs_pop_add(gs, rg1, parent = "root")## [1] 2recompute(gs)
gs_get_pop_paths(gs)## [1] "root" "/NoneDebris"ggcyto(fs_comp_trans[[1]], aes(x="FSC.A", y="SSC.A")) +
geom_hex(bins=256) +
geom_gate(gs_pop_get_gate(gs, "NoneDebris")) +
theme_classic()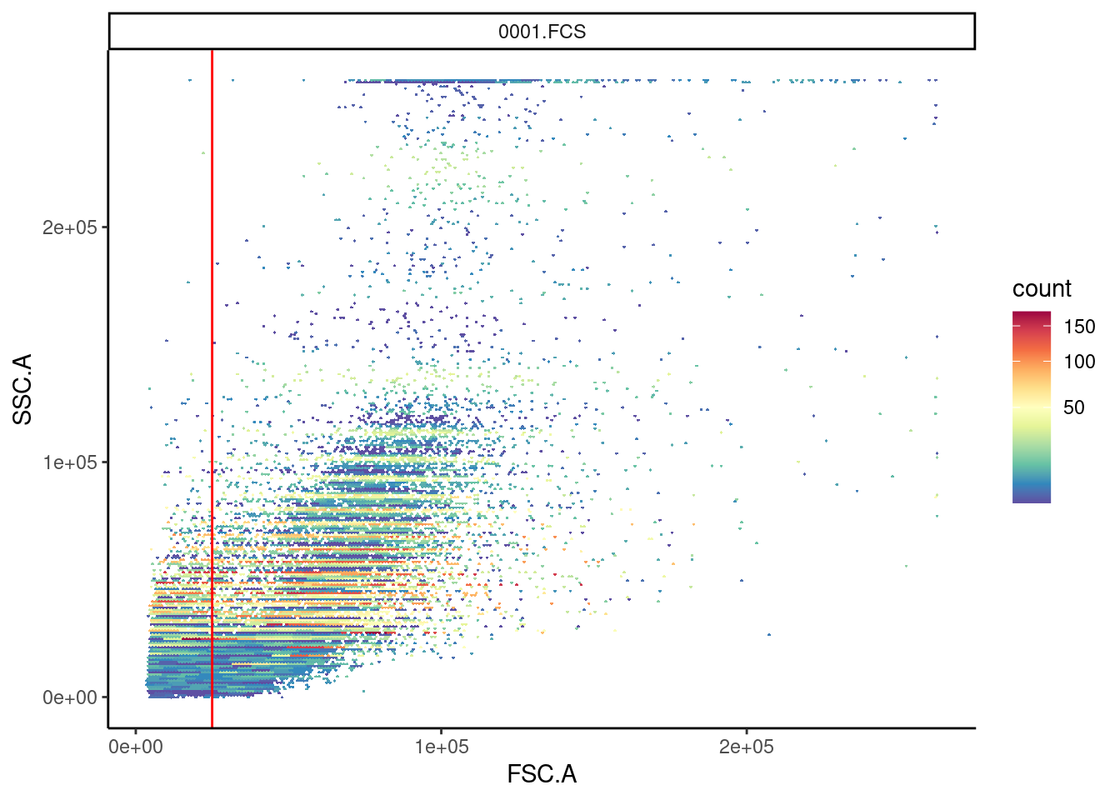
gs_pop_get_stats(gs)## sample pop count
## 1: 0001.FCS root 500000
## 2: 0001.FCS /NoneDebris 361522
## 3: 0002.FCS root 487971
## 4: 0002.FCS /NoneDebris 400537
## 5: 0003.FCS root 401831
## 6: 0003.FCS /NoneDebris 287182其中存在一些问题,在于若是设置了范围为25000-无穷,则有一些问题在于在下图左上角的图不知道准确数值,于是修改代码改的更智能。
thisData <- gs_pop_get_data(gs)
nonDebris_gate <- fsApply(thisData, function(fr) openCyto:::.mindensity(fr, channels = "FSC.A"))
gs_pop_add(gs, nonDebris_gate, parent = "root", name = "nonDebris")## [1] 3recompute(gs)
ggcyto(fs_comp_trans[[1]], aes(x="FSC.A", y="SSC.A")) +
geom_hex(bins=256) +
geom_gate(gs_pop_get_gate(gs, "NoneDebris")) +
theme_classic()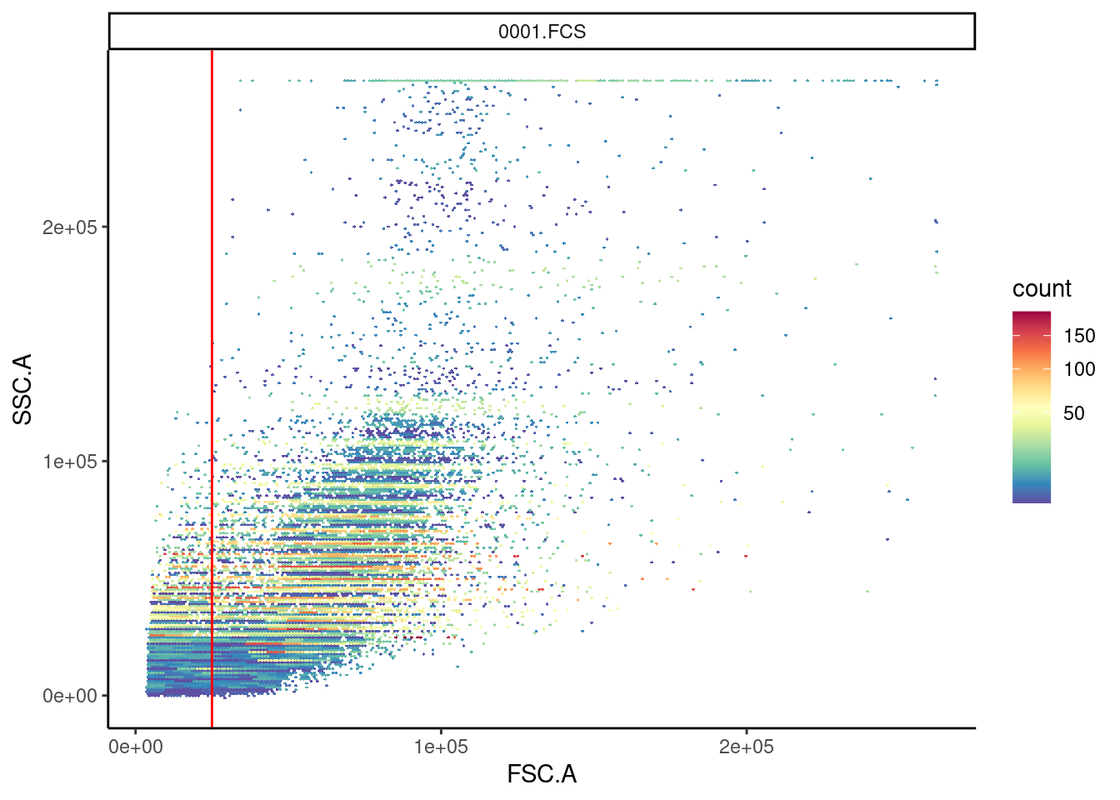
在之前的介绍中,已经基本上完成了流式依赖包的大部分也是主要功能。由于R语言的绘图以及自动化功能非常强大,所以正常而言在flowCore、openCyto和ggcyto依赖包解决不了的问题可以通过ggplot2进行解决。
出现了一个getData()的命令,而在新版本的R语言中,该命令已经不存在了,用一下代码进行替换。
其中非常重要的一环在于如果没有这个命令,后续建立门的操作就都无效了,这条命令的目的就是调用文件中的数据。如果调用失败,当然会报错了。
在绘制流式散点图的时候,可以通过以下命令不但绘制出散点图,而且还overlay了一个之前建立好的椭圆型范围的门。
ggcyto(fs_comp_clean_trans, aes(x="FITC.A",y="PE.A")) +
geom_hex(bins=256) +
geom_gate(gs_pop_get_gate(gs, "flowflust")) +
theme_classic()而绘制密度图,则代码如下:
ggcyto(fs_comp_clean_trans[[1]], aes(x="FITC.A")) +
geom_density(fill="lightgrey", alpha=0.3) +
geom_overlay(fs_comp_clean_tran[[2]], fill="lightblue", alpha=0.3) +
geom_overlay(fs_comp_clean_tran[[3]], fill="lightgren", alpha=0.3) +
facet_null() +
theme_classic()而上述代码所绘制出的图有一个问题在于无法加注图例,因此我的改进方法如下:
data1 <- fs_comp_clean_trans[[1]]@exprs
data2 <- fs_comp_clean_trans[[2]]@exprs
data3 <- fs_comp_clean_trans[[3]]@exprs
data1 <- as.data.frame(data1)
data2 <- as.data.frame(data2)
data3 <- as.data.frame(data3)
data1$Type <- "Blank"
data2$Type <- "FITC-A"
data3$Type <- "FITC-A+"
data <- rbind(data1, data2, data3)
##开始绘图
ggplot(data, aes(x=data$FITC.A, fill=data$Type)) +
geom_density(alpha=0.3) +
scale_fill_manual(values=c("lightgrey","lightblue","lighgreen")) +
scale_x_logicle() +
theme_classic() +
theme(legend.position=c(0.8,0.5), legnd.title=element_blank())在后期的应用中又发现了一些问题,并在此进行总结。
autoplot(fs_comp_clean_trans[[1]], x="FITC.A",y="PE.A", bins=256) +
theme_classic() +
ggcyto_par_set(limits="instrument") +
scale_x_flowjo_biexp() +
scale_y_flowjo_biexp() +
# geom_vline(xintercept = 451.7652) +
# geom_hline(yintercept = 27258.43) +
geom_gate(flust.c, colour="black") +
geom_stats(type = "count", hjust=-0.5, fill=NA) +
geom_text(x=1500,y=1000,label="Lymph")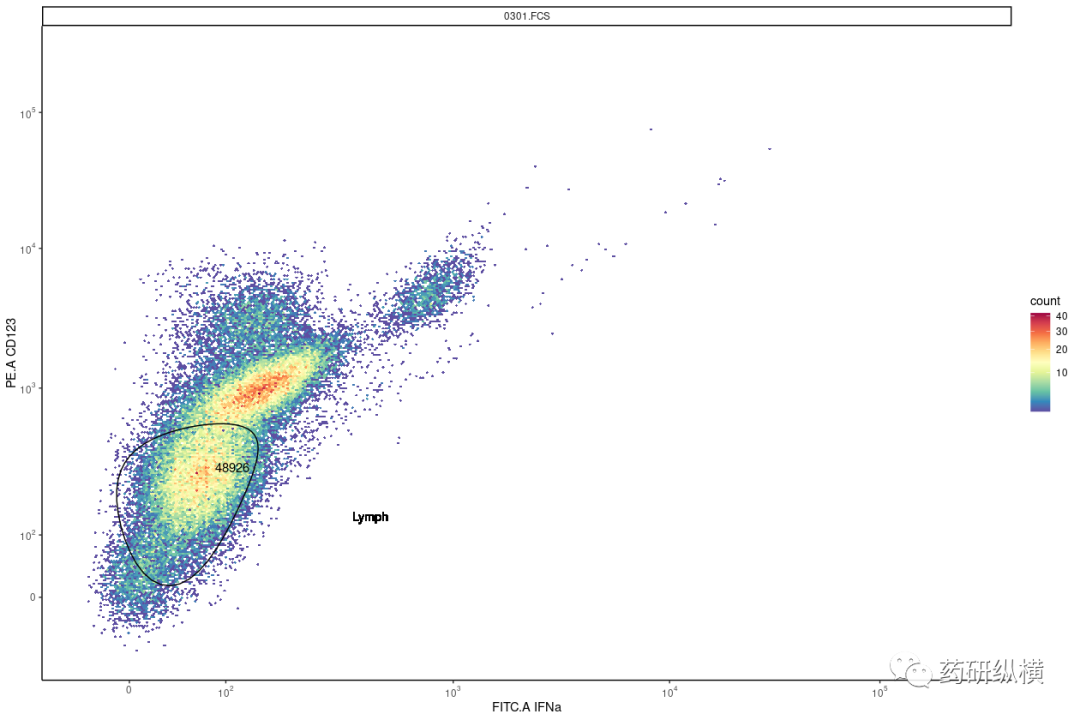
我们已知FSC和SSC能够进行细胞的分选并用gate进行框选分析。但是应用的过程中,小编发现在流式技术中最经常使用的十字象限并不能有效的显示出来,因为quadGate()的命令和科学计数法的坐标轴不兼容,在坐标轴不为科学计数法的时候,十字象限显示正常,而坐标轴转换成科学计数法时,则不显示十字象限。因此,小编尝试了解决方法并上网冲浪寻找解决办法,但是可惜的是并没有找到有效的解决方法。那么小编采用了土办法,就是在ggplot框架中加了两条直线:geom_vline()和geom_hline()就成功的解决了这个问题(说的很简单,过程并不容易,中间通过openCyto:::.mindensity()的命令自动计算出了数值,并将这个数值输入到vline和hline两条命令里)。并且在调用象限门时,还可以通过geom_stats()命令在四个象限自动显示细胞数/百分比。
另外加之,在之前的加门的教程中,由于应用的并不多,所以在叙述中简单的带过,在应用中小编用的最多的就是椭圆形的门进行叙述。
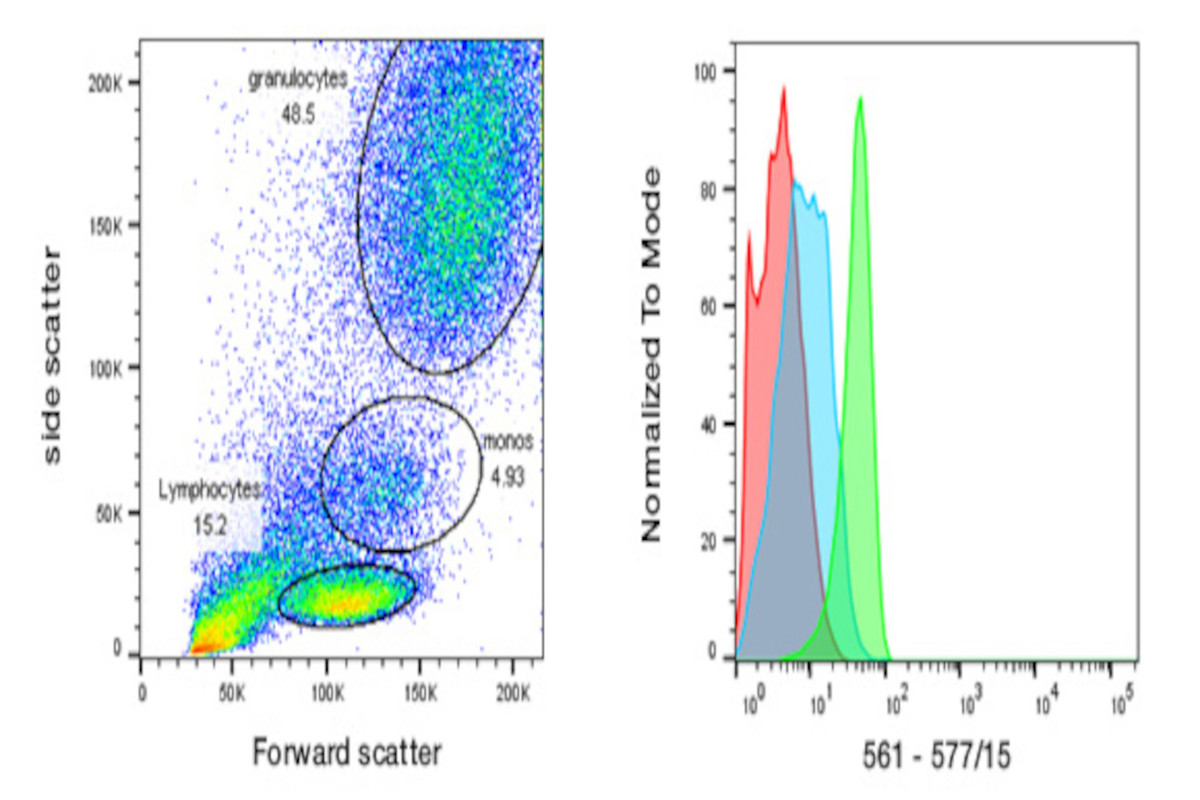
首先以FSC.A为横坐标,SSC.A为纵坐标进行绘制,并框选出需要的位置即中心的坐标值。
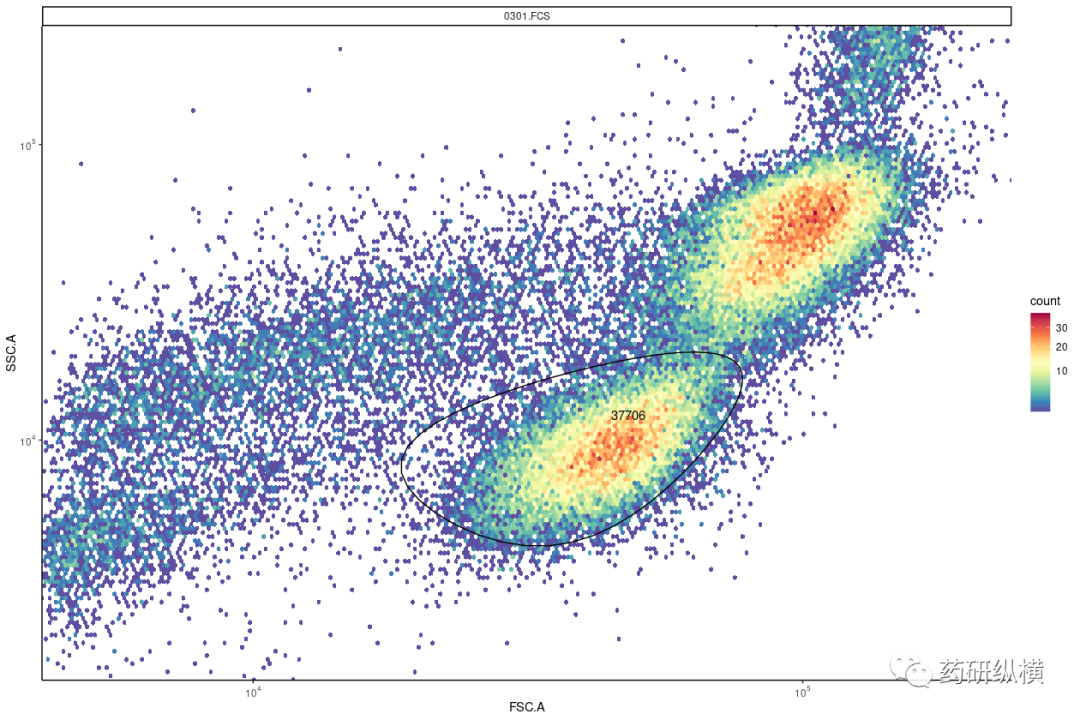
并用所用的荧光通道对SSC.A进行作图,确定在FITC.A中所需要的位置,并用FITC.A对PE.A作图,如图一所示。
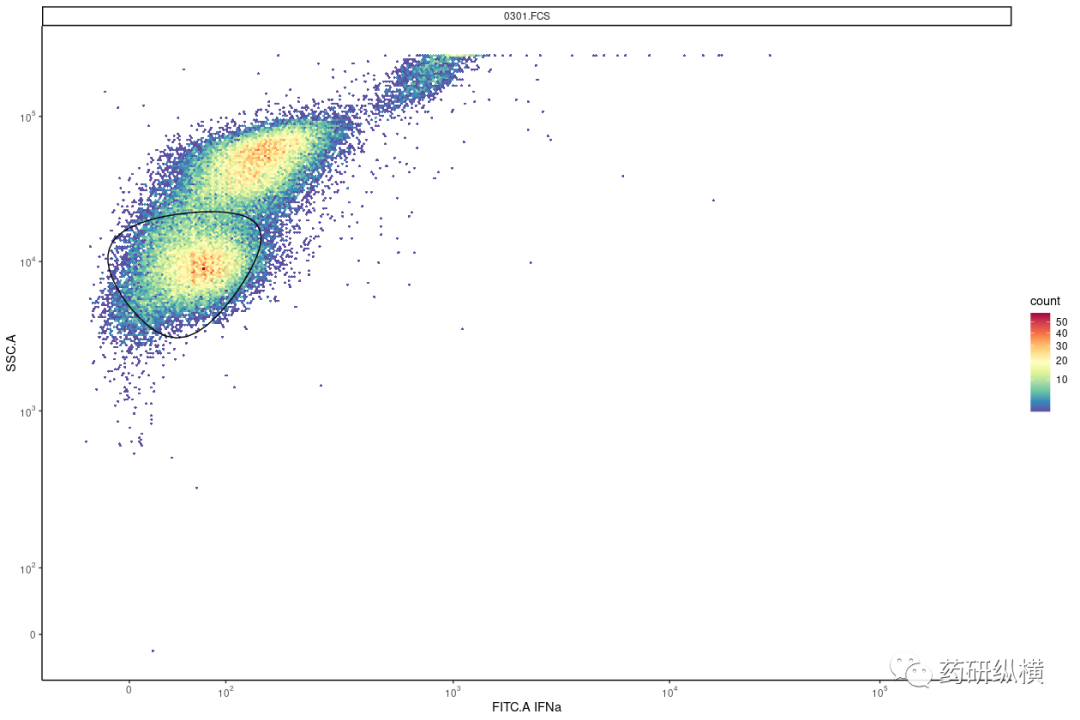
autoplot(auto_gs[1], "FITC.A", "PE.A", gate="flust.fitc.pe", bins=156) +
scale_x_flowjo_biexp() +
scale_y_flowjo_biexp() +
theme_classic()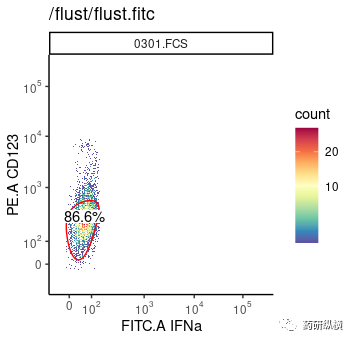
通过上述命令可以只显示门内的流式散点图。但是在上述代码中需要注意的是在所有的教程中都没有书籍,在autoplot()里一定要加gate=““,如果不加就会报错,而网上所有的教程都没有加,这有可能是由于R及依赖包版本更新的问题。
{kind=link}